High-Entropy Ceramics
High-entropy ceramics [1] are an emerging research field of materials that combine the stabilizing effects of high-configurational entropy with metal-nonmetal bonding, offering promising candidates for a variety of applications ranging from hypersonics to batteries. Due to the limits of density functional theory, modeling the prominent chemical disorder in these systems is a great challenge. Our group has developed the Partial OCCupation (POCC) [2] algorithm to describe disordered materials using a series of small supercells and thermodynamic averaging. The approach has been used to develop entropy descriptors [3], such as the entropy-forming ability (EFA), that reliably predict the synthesizability of high-entropy materials. This approach led to the discovery of new high-entropy carbides [3], and can greatly accelerate the discovery of new disordered materials when coupled with machine learning [4]. Our current research focuses on the prediction of new high-entropy ceramics and their physical properties using the POCC approach.

- C. Oses, C. Toher, and S. Curtarolo, High-entropy ceramics, Nat. Rev. Mater. 5, 295-309 (2020) doi.org/10.1038/s41578-019-0170-8
- K. Yang, C. Oses, and S. Curtarolo, Modeling off-stoichiometry materials with a high-throughput ab-initio approach, Chem. Mater. 28(18), 6484-6492 (2016). doi.org/10.1021/acs.chemmater.6b01449
- P. Sarker, T.J. Harrington, C. Toher, C. Oses, M. Samiee, J.-P. Maria, D.W. Brenner, K.S. Vecchio, and S. Curtarolo, High-entropy high-hardness metal carbides discovered by entropy descriptors, Nature Communications 9, 4980 (2018). doi.org/10.1038/s41467-018-07160-7
- K. Kaufmann, D. Maryanovsky, W.M. Mellor, C. Zhu, A.S. Rosengarten, T.J. Harrington, C. Oses, C. Toher, S. Curtarolo, and K.S. Vecchio, Discovery of novel high-entropy ceramics via machine learning, npj Comput. Mater. 6, 42 (2020). doi.org/10.1038/s41524-020-0317-6
Phase Stability
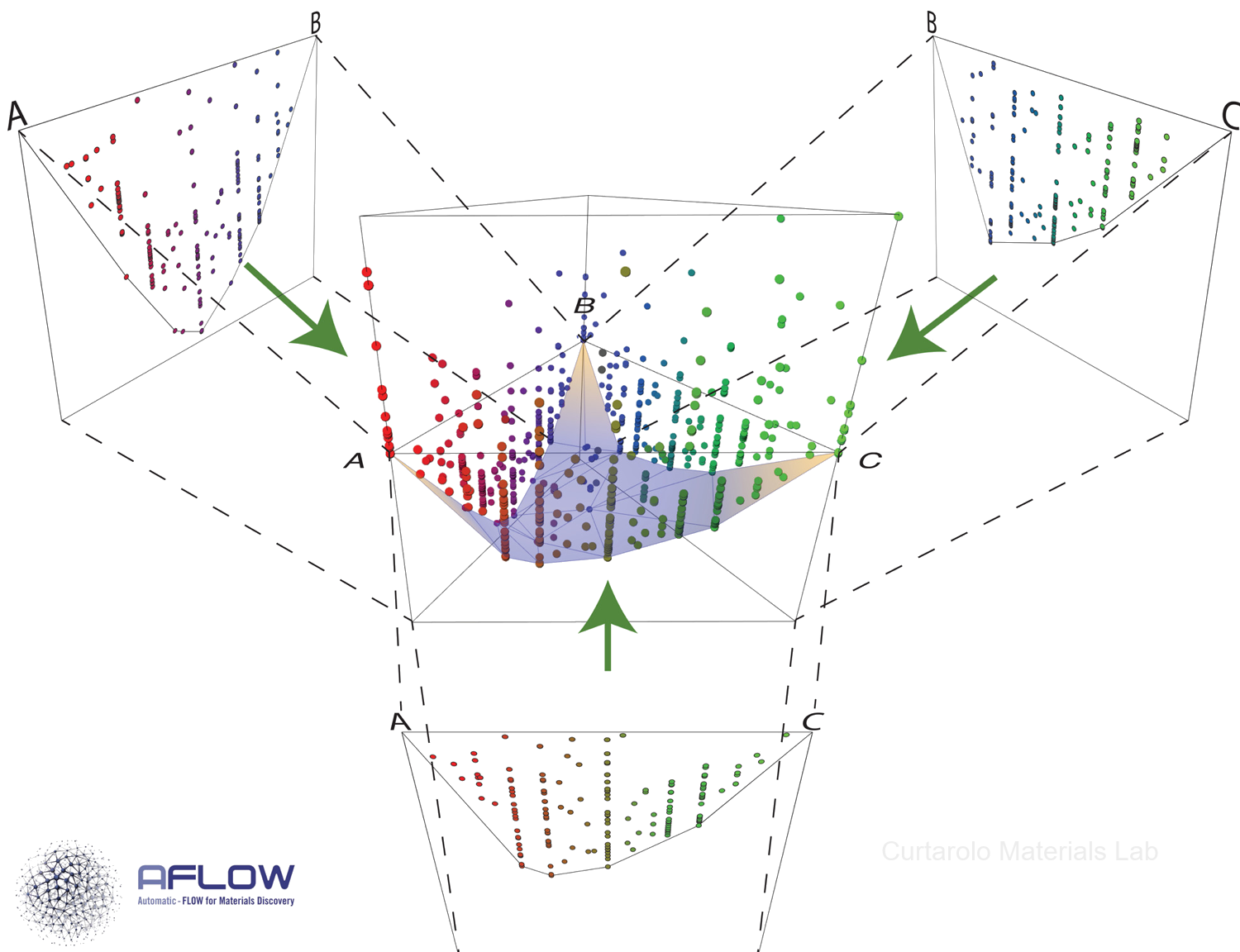
The discovery of new functional materials demands an efficient determination of the thermodynamically stable — and synthesizable — phases. In our group, we focus on predicting the phase stability of novel materials using a combination of density functional theory calculations [1], first-principles thermodynamics (LTVC method) [2], entropy descriptors [3], and machine learning methods [4]. In addition, our group has created the Automatic FLOW for Materials Discovery (AFLOW) software, a framework for high-throughput materials discovery. The modules AFLOW-CHULL [5] and AFLOW-CCE [6] have been developed to perform thermodynamic stability analysis and to correct DFT formation enthalpies of polar materials based on local atomic environments, respectively.
- S. Barzilai, C. Toher, S. Curtarolo, and O. Levy, The molybdenum-titanium phase diagram evaluated from ab-initio calculations, Phys. Rev. Materials 1, 023604 (2017). doi.org/10.1103/PhysRevMaterials.1.023604
- Y. Lederer, C. Toher, K.S. Vecchio, and S. Curtarolo, The search for high-entropy alloys: a high-throughput ab-initio approach, Acta Mater. 159, 364-383 (2018). doi.org/10.1016/j.actamat.2018.07.042
- P. Sarker, T.J. Harrington, C. Toher, C. Oses, M. Samiee, J.-P. Maria, D.W. Brenner, K.S. Vecchio, and S. Curtarolo, High-entropy high-hardness metal carbides discovered by entropy descriptors, Nature Communications 9, 4980 (2018). doi.org/10.1038/s41467-018-07160-7
- R. Ouyang, S. Curtarolo, E. Ahmetcik, M. Scheffler, and L.M. Ghiringhelli, SISSO: a compressed-sensing method for identifying the best low-dimensional descriptor in an immensity of offered candidates, Phys. Rev. Materials 2, 083802 (2018). doi.org/10.1103/PhysRevMaterials.2.083802
- C. Oses, E. Gossett, D. Hicks, F. Rose, M.J. Mehl, E. Perim, I. Takeuchi, S. Sanvito, M. Scheffler, Y. Lederer, O. Levy, C. Toher, and S. Curtarolo, AFLOW-CHULL: Cloud-oriented platform for autonomous phase stability analysis, J. Chem. Inf. Model. 58(12), 2477-2490 (2018). doi.org/10.1021/acs.jcim.8b00393
- R. Friedrich, D. Usanmaz, C. Oses, A.R. Supka, M. Fornari, M. Buongiorno Nardelli, C. Toher, and S. Curtarolo, Coordination corrected ab initio formation enthalpies, npj Comput. Mater. 5, 59 (2019). doi.org/10.1038/s41524-019-0192-1
Symmetry and Prototyping
AFLOW features a wide variety of structural analysis tools to aid in automatic materials discovery. The AFLOW-SYM module [1] calculates a
complete suite of crystallographic symmetries, including different symmetry groups, Wyckoff positions, and lattice types (real, reciprocal, and superlattice). Self-consistent routines and an adaptive
tolerance scheme guarantee the results will be commensurate with crystallographic conventions. Compared to other symmetry packages, AFLOW-SYM is more consistent with experimentally resolved symmetries without
the need for users to tune tolerance thresholds.
The AFLOW Prototype Encyclopedia is a collection of unique crystalline prototype structures that can be automatically generated with the
AFLOW software. The encyclopedia features over 1,100 structure prototypes — cataloged across Part 1 [2],
Part 2 [3], and Part 3 [4] — and is continuing to
grow. Each prototype is classified by an AFLOW prototype label and its degrees of freedom.
AFLOW-XtalFinder [5] identifies and classifies unique structural prototypes. The module symbolically maps structures into their ideal
prototype designation and internal degrees of freedom consistent with the International Tables for Crystallography. This functionality enables the AFLOW database to be searched by structure-type. Built-in
methods also establish uniqueness/equivalency with respect to users’ input structures, the aflow.org repository, and the
AFLOW Prototype Encyclopedia.

- D. Hicks, C. Oses, R.H. Taylor, E. Gossett, G. Gomez, C. Toher, M.J. Mehl, O. Levy, and S. Curtarolo, AFLOW-SYM: Platform for the complete, automatic and self-consistent symmetry analysis of crystals, Acta Cryst. A. 74, 184-203 (2018). doi.org/10.1107/S2053273318003066
- M.J. Mehl, D. Hicks, C. Toher, O. Levy, R.M. Hanson, G.L.W. Hart, and S. Curtarolo, The AFLOW Library of Crystallographic Prototypes: Part 1, Comp. Mat. Sci. 136(Supplement), S1-S828 (2017). doi.org/10.1016/j.commatsci.2017.01.017
- D. Hicks, M.J. Mehl, E. Gossett, C. Toher, O. Levy, R.M. Hanson, G.L.W. Hart, and S. Curtarolo, The AFLOW Library of Crystallographic Prototypes: Part 2, Comp. Mat. Sci. 161(Supplement), S1-S1011 (2019). doi.org/10.1016/j.commatsci.2018.10.043
- D. Hicks, M.J. Mehl, M. Esters, C. Oses, O. Levy, G.L.W. Hart, C. Toher, and S. Curtarolo, The AFLOW Library of Crystallographic Prototypes: Part 3, Comp. Mat. Sci. 199, 110450 (2021). doi.org/10.1016/j.commatsci.2021.110450
- D. Hicks, C. Toher, D.C. Ford, F. Rose, C. De Santo, O. Levy, M.J. Mehl, and S. Curtarolo, AFLOW-XtalFinder: a reliable choice to identify crystalline prototypes, npj Comput. Mater. 7, 30 (2021). doi.org/10.1038/s41524-020-00483-4
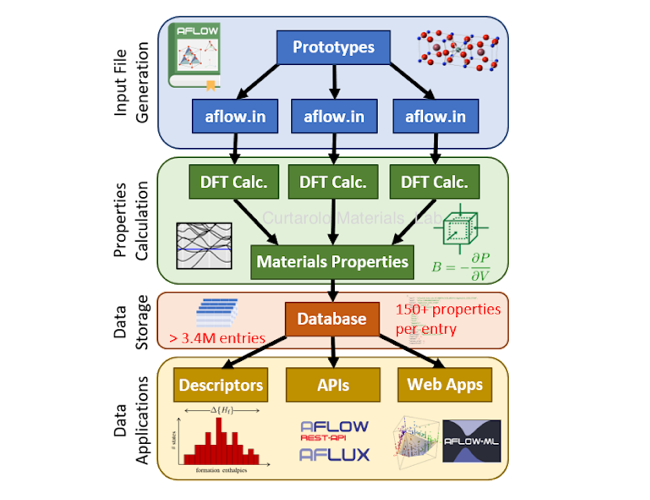
Autonomous Property Prediction
Our group has developed the Automatic FLOW for Materials Discovery (AFLOW) software, a framework for high-throughput autonomous materials properties predictions.
Through standardized[1] density functional theory calculations, we have created the biggest, publicly available materials
databases in the world, and provide APIs to search[2] and
access[3] the data.
Modules have been developed to calculate electronic structures[4], thermomechanical (AFLOW-AEL [5] and AFLOW-AGL[6])
and vibrational (AFLOW-APL [7]
and AFLOW-QHA [8]) properties and lattice
thermal conductivities (AFLOW-AAPL [9]). We have further implemented
machine learning [10] algorithms to predict thermomechanical and vibrational properties.
- C.E. Calderon, J.J. Plata, C. Toher, C. Oses, O. Levy, M. Fornari, A. Natan, M.J. Mehl, G.L.W. Hart, M. Buongiorno Nardelli, and S. Curtarolo, The AFLOW Standard for High-Throughput Materials Science Calculations, Comput. Mater. Sci., 108(Part A), 233-238 (2015). doi.org/10.1016/j.commatsci.2015.07.019
- F. Rose, C. Toher, E. Gossett, C. Oses, M. Buongiorno Nardelli, M. Fornari, and S. Curtarolo, AFLUX: The LUX materials search API for the AFLOW data repositories, Comp. Mat. Sci. 137, 362-370 (2017). doi.org/10.1016/j.commatsci.2017.04.036
- R.H. Taylor, F. Rose, C. Toher, O. Levy, K. Yang, M. Buongiorno Nardelli, and S. Curtarolo, A RESTful API for exchanging materials data in the AFLOWLIB.org consortium, Comp. Mat. Sci. 93, 178-192 (2014). doi.org/10.1016/j.commatsci.2014.05.014
- W. Setyawan and S. Curtarolo, High-throughput electronic band structure calculations: challenges and tools, Comp. Mat. Sci. 49, 299-312 (2010). doi.org/10.1016/j.commatsci.2010.05.010
- C. Toher, C. Oses, J.J. Plata, D. Hicks, F. Rose, O. Levy, M. de Jong, M.D. Asta, M. Fornari, M. Buongiorno Nardelli, and S. Curtarolo, Combining the AFLOW GIBBS and Elastic Libraries for efficiently and robustly screening thermo-mechanical properties of solids, Phys. Rev. Materials 1, 015401 (2017). doi.org/10.1103/PhysRevMaterials.1.015401
- C. Toher, J.J. Plata, O. Levy, M. de Jong, M.D. Asta, M. Buongiorno Nardelli, and S. Curtarolo, High-throughput computational screening of thermal conductivity, Debye temperature, and Grüneisen parameter using a quasiharmonic Debye model, Phys. Rev. B 90, 174107 (2014). doi.org/10.1103/PhysRevB.90.174107
- M. Esters, C. Oses, D. Hicks, M.J. Mehl, M. Jahnátek, M.D. Hossain, J.-P. Maria, D.W. Brenner, C. Toher, and S. Curtarolo, Settling the matter of the role of vibrations in the stability of high-entropy carbides, Nat. Commun., 12, 5747 (2021). doi.org/10.1038/s41467-021-25979-5
- P. Nath, D. Usanmaz, D. Hicks, C. Oses, M. Fornari, M. Buongiorno Nardelli, C. Toher, and S. Curtarolo, AFLOW-QHA3P: Robust and automated method to compute thermodynamic properties of solids, Phys. Rev. Materials 3, 073801 (2019). doi.org/10.1103/PhysRevMaterials.3.073801
- J.J. Plata, P. Nath, D. Usanmaz, J. Carrete, C. Toher, M. de Jong, M.D. Asta, M. Fornari, M. Buongiorno Nardelli, and S. Curtarolo, An efficient and accurate framework for calculating lattice thermal conductivity of solids: AAPL - AFLOW Anharmonic Automatic Phonon Library, npj Comput. Mater. 3, 45 (2017). doi.org/10.1038/s41524-017-0046-7
- E. Gossett, C. Toher, C. Oses, O. Isayev, F. Legrain, F. Rose, E. Zurek, J. Carrete, N. Mingo, A. Tropsha, and S. Curtarolo, AFLOW-ML: A RESTful API for machine-learning predictions of materials properties, Comp. Mat. Sci. 152, 134-145 (2018). doi.org/10.1016/j.commatsci.2014.05.014
Metallic Glasses
Metallic glasses are a unique class of materials whose defining characteristic is the absence of the crystalline order typically found in metals. While most metals can be transformed into a glassy state from
a melt when cooled quickly, generating them as a bulk material is challenging. The vast phase space creates challenges for experimental exploration, necessitating complementary in silico contributions
to prioritize promising materials.
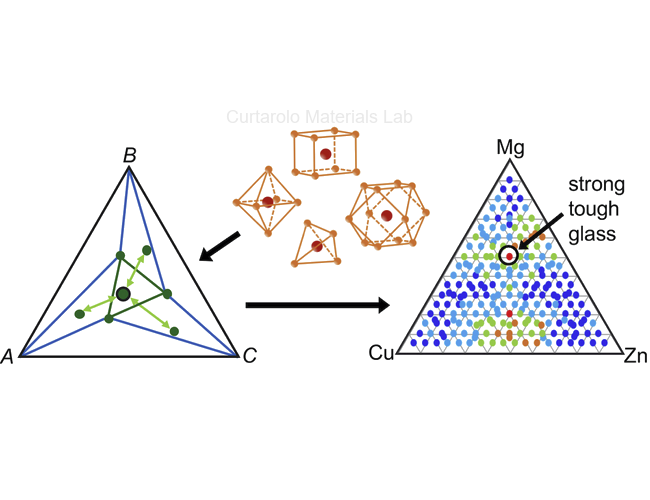
Our approach to predicting the Glass Forming Ability (GFA) is based on quantifying the competition of initially formed crystal nuclei in the rapidly cooled material. Our group first introduced a
spectral descriptor for GFA [1] for binary alloys. Later, we extended the concept to alloys with three elements and incorporated
off-stoichiometric contributions [2]. Local atomic environments form the basis for these structural comparisons. As the systems
become more complex by including more elements, developing robust atomic environment methods that work reliably for non-ideal geometries is one of our priorities. Drawing from the AFLOW material database, the
GFA of new systems can be calculated efficiently, helping to prioritize the most promising material combinations.
- E. Perim, D. Lee, Y. Liu, C. Toher, P. Gong, Y. Li, W.N. Simmons, O. Levy, J.J. Vlassak, J. Schroers, and S. Curtarolo, Spectral descriptors for bulk metallic glasses based on the thermodynamics of competing crystalline phases, Nature Communications 7, 12315 (2016). doi.org/10.1038/ncomms12315
- D.C. Ford, D. Hicks, C. Oses, C. Toher, and S. Curtarolo, Metallic glasses for biodegradable implants, Acta Mater. 176, 297-305 (2019). doi.org/10.1016/j.actamat.2019.07.008